-
神經系統疾病新機制:線粒體轉移功能障礙
發布時間: 2026-03-24 點擊次數: 72次《LRRK2 G2019S突變通過Drp1-STX17依賴的方式導致線粒體轉移功能障礙》LRRK2 G2019S突變通過增強Drp1 Ser616磷酸化,導致STX17從線粒體脫落,從而損害星形膠質細胞向多巴胺能神經元的線粒體轉移功能,而抑制Drp1磷酸化的DUSP6可恢復這一過程并發揮神經保護作用。成果發表在Translational Neurodegeneration雜志(IF:15.2);
《Translational Neurodegeneration》是一本專注于神經退行性疾病領域的開放獲取、同行評議的國際英文學術期刊。期刊創刊于2012年1月,由上海交通大學醫學院附屬瑞金醫院神經科、神jing病學研究所與施普林格·自然集團合作創辦。涵蓋所有神經退行性疾病的相關研究,包括但不限于帕金森病及運動障礙、阿爾茨海默病及其他癡呆、運動神經元病等。感興趣的主題包括流行病學、病原學、診斷標志物、新藥開發、治療及預防等。影響因子為15.2,5年影響因子為15.0;由BioMed Central出版,電子國際標準連續出版物號為 2047-9158;
研究背景:
帕金森病是僅次于阿爾茨海默病的第二大神經退行性疾病,其主要病理特征是中腦黑質區多巴胺能神經元的進行性喪失。盡管目前以多巴胺替代療法為主的對癥治療能夠改善癥狀,但無法阻止或延緩疾病的進展,因此探索能夠抑制神經元凋亡的內源性神經保護機制具有重要意義。遺傳因素和環境因素共同參與了帕金森病的發生。其中,LRRK2 G2019S突變是zui常見的帕金森病致病基因變異,可顯著增強LRRK2激酶活性,導致線粒體自噬障礙、線粒體DNA損傷和線粒體分裂異常等病理改變。同時,前期研究已證實,星形膠質細胞可以將健康的線粒體轉移給受損的多巴胺能神經元,這可能是腦內一種內源性的神經修復機制。然而,在LRRK2 G2019S突變背景下,這種線粒體轉移功能是否受損,以及其具體分子機制尚不清楚。
研究方法:
研究采用誘導多能干細胞技術,從健康個體和攜帶LRRK2 G2019S突變的帕金森病患者外周血中成功誘導分化獲得多巴胺能神經元和星形膠質細胞,建立了模擬腦內微環境的“星形膠質細胞-神經元"共培養體系。通過魚藤酮處理模擬環境毒素暴露,實驗首先觀察了不同基因背景下線粒體轉移效率的變化;進而利用MitoTracker標記星形膠質細胞線粒體,結合激光共聚焦顯微鏡和流式細胞術,定量檢測共培養體系中神經元內星形膠質細胞來源的線粒體數量,并測定神經元ATP水平以評估其功能狀態。在分子機制探討層面,通過siRNA技術分別敲低SNARE家族蛋白STX17、VAMP3和SNAP23,篩選影響線粒體釋放的關鍵蛋白;運用免疫熒光共定位分析STX17與線粒體外膜蛋白TOM20的共定位關系;采用免疫共沉淀技術驗證Drp1與STX17的相互作用;并通過Western blot檢測不同處理下Drp1總蛋白及其Ser616位點磷酸化水平的變化。最后,使用Drp1磷酸化抑制劑DUSP6進行功能挽救實驗,觀察其對STX17線粒體定位、線粒體轉移效率及神經元樹突損傷的改善作用。
主要研究結果:
1. iPSCs誘導分化為星形膠質細胞和多巴胺能神經元
iPSCs來源于健康供體和攜帶LRRK2 G2019S雜合突變帕金森病患者的PBMCs,該患者是我團隊報道的中國shou例攜帶LRRK2 G2019S突變的帕金森病病例[31]。iPSCs的核型分析顯示正常的46XY核型,無染色體異常。最初,使用雙重抑制劑SB431542和Noggin抑制SMAD信號通路,誘導iPSCs神經化為NSCs。通過向培養體系中添加各種小分子,成功實現了向星形膠質細胞和多巴胺能神經元的分化。并通過Western blot和免疫熒光對兩種細胞的標志物進行驗證。
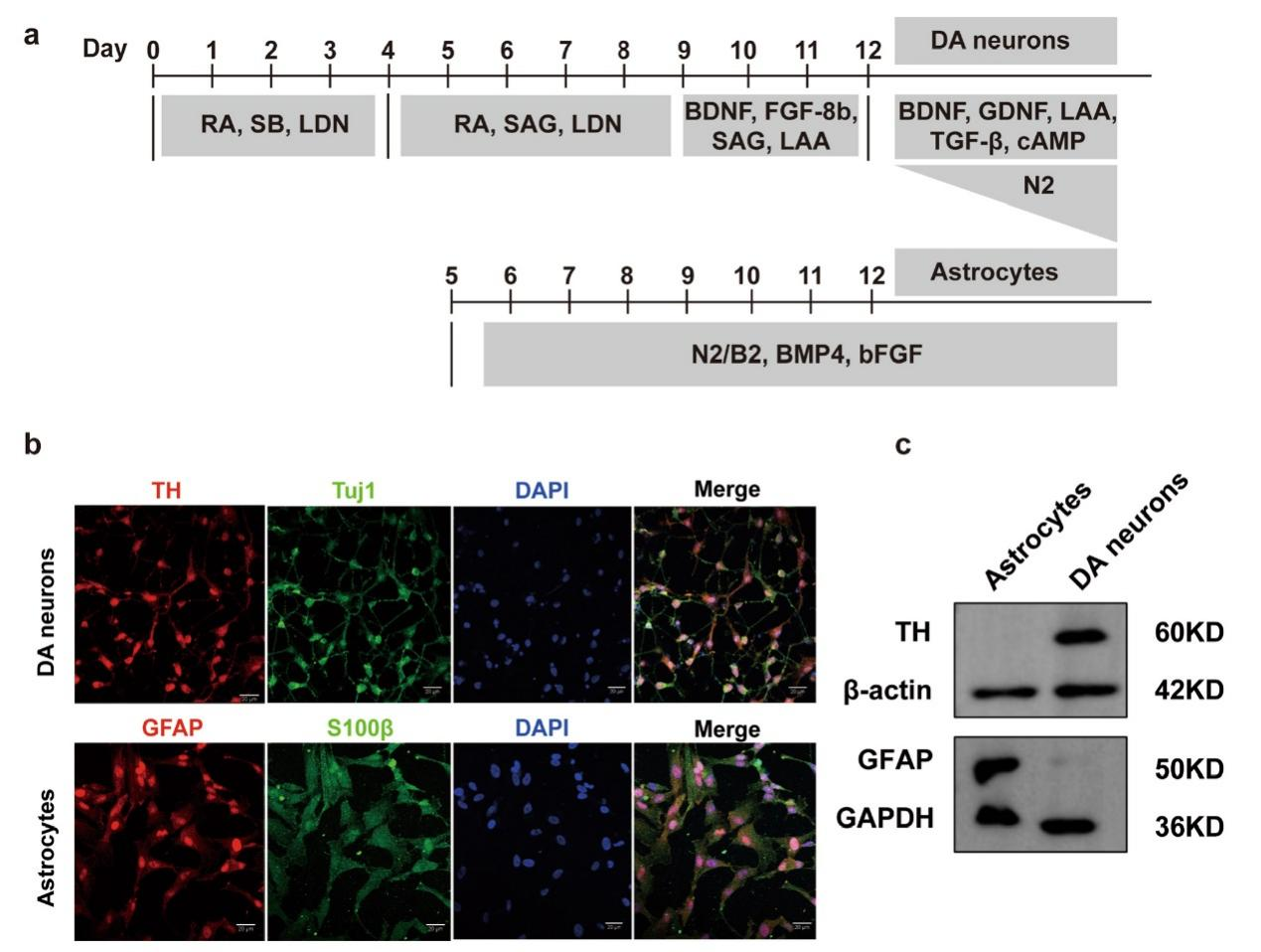
圖1,人iPSC來源的多巴胺能神經元和星形膠質細胞的誘導
2. LRRK2 G2019S突變減少線粒體從星形膠質細胞向多巴胺能神經元的轉移
在突變型共培養體系(突變星形膠質細胞+突變神經元)中,200 nM魚藤酮處理后神經元樹突長度減少約60%,損傷程度顯著重于野生型共培養體系(減少44%),表明突變神經元對環境毒素的抵抗力降低。當野生型星形膠質細胞與突變神經元共培養時,樹突長度減少約50%,損傷輕于突變共培養體系,提示健康星形膠質細胞能部分緩解突變神經元的損傷。MitoTracker線粒體示蹤顯示,魚藤酮呈劑量依賴性地減少神經元內星形膠質細胞來源的線粒體數量,突變體系中線粒體數量減少70%,野生型減少60%,說明突變導致線粒體轉移功能受損更嚴重。流式細胞術檢測星形膠質細胞條件培養基證實,突變星形膠質細胞釋放線粒體的能力下降更明顯。此外,將魚藤酮處理后的條件培養基與神經元共培養發現,突變神經元ATP水平下降更顯著,表明線粒體轉移功能受損導致神經元能量供應不足。這些結果共同揭示了LRRK2 G2019S突變通過損害線粒體轉移增強神經元對環境毒素的敏感性。
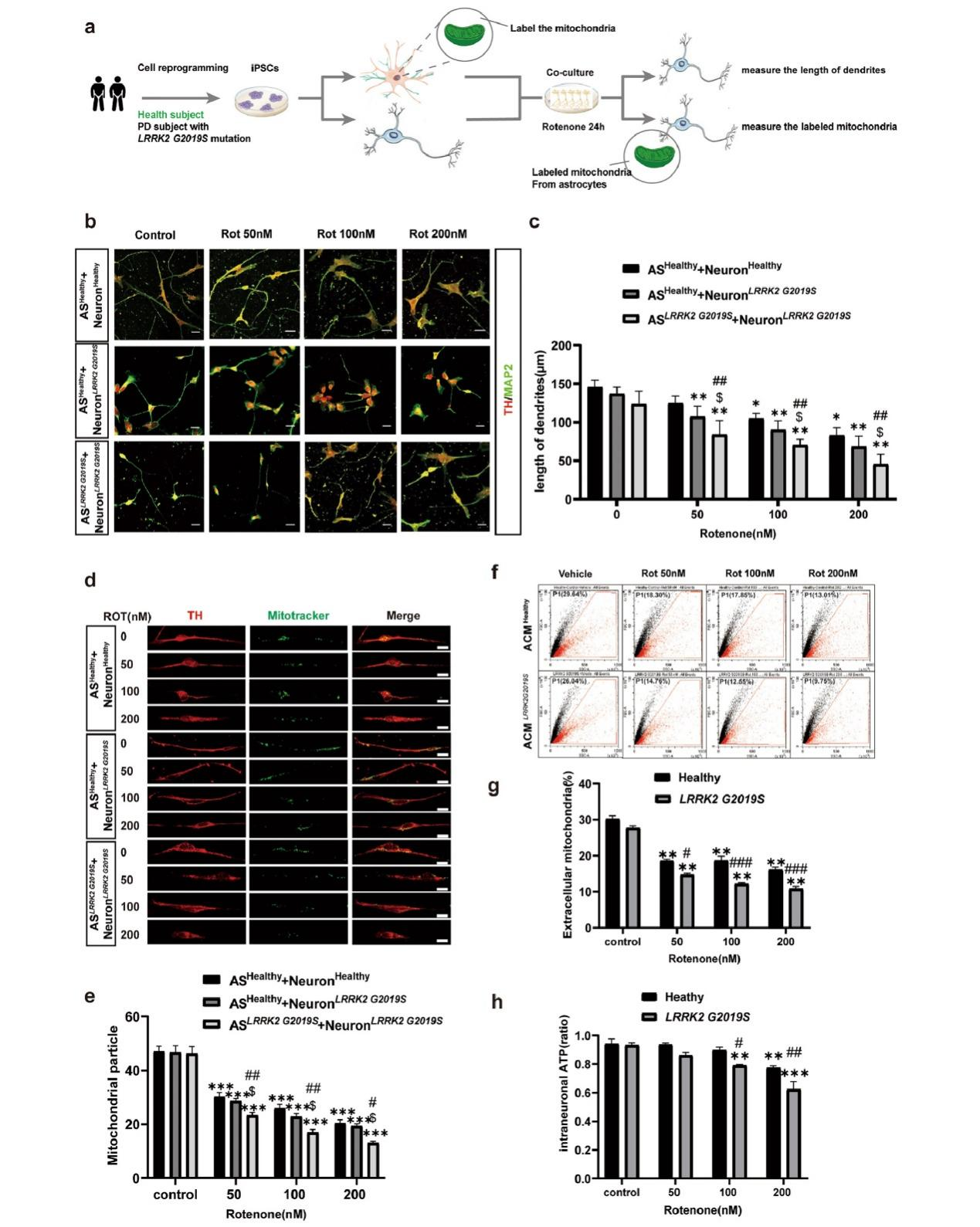
圖2,LRRK2 G2019S突變與魚藤酮聯合作用導致多巴胺能神經元變性,并損害線粒體從星形膠質細胞向多巴胺能神經元的轉移
3. 膜融合相關蛋白STX17通過不依賴SNARE的機制參與線粒體輸出
通過siRNA分別敲低STX17、VAMP3和SNAP23后發現,STX17敲低使星形膠質細胞條件培養基中線粒體數量減少70.70%,而VAMP3和SNAP23敲低僅減少約27%,表明STX17是線粒體釋放的關鍵調控蛋白。免疫共沉淀顯示VAMP3與STX17、SNAP23均無結合,免疫熒光進一步證實STX17與線粒體外膜蛋白TOM20共定位,而VAMP3和SNAP23不與TOM20共定位,證明STX17的作用不依賴經典SNARE復合物形成。在魚藤酮處理的星形膠質細胞中,STX17與TOM20的共定位程度隨魚藤酮濃度升高而降低,且突變型星形膠質細胞中這種下降更為顯著。值得注意的是,線粒體標志物TOM20和VDAC1的蛋白水平在不同基因型和不同濃度魚藤酮處理下均無顯著變化,表明線粒體數量保持穩定,排除了線粒體自噬或降解對共定位結果的影響。然而,200 nM魚藤酮使野生型星形膠質細胞STX17熒光強度降低27%,突變型降低41%,Western blot驗證了這一結果。進一步的共定位分析證實,STX17定位于線粒體外膜的能力下降是導致線粒體轉移功能受損的直接原因。這些結果揭示了STX17在線粒體釋放中的關鍵作用及其受突變和環境毒素協同調控的機制。
圖3的核心研究發現是STX17通過不依賴經典SNARE復合物的機制參與星形膠質細胞線粒體釋放,且LRRK2 G2019S突變與魚藤酮協同損害STX17的線粒體定位。

圖3,STX17通過不依賴SNARE的機制參與星形膠質細胞的線粒體輸出
4. 線粒體Drp1在線粒體轉移中發揮作用
通過siRNA敲低Drp1后,星形膠質細胞條件培養基中線粒體數量顯著減少,共培養體系中神經元內星形膠質細胞來源的線粒體也明顯減少,證實Drp1是線粒體釋放的關鍵調控蛋白。免疫共沉淀結果顯示,Drp1與STX17存在直接相互作用,為Drp1調控STX17功能提供了分子基礎。進一步檢測發現,隨著魚藤酮濃度升高,Drp1 Ser616位點磷酸化水平呈劑量依賴性增加,且突變型星形膠質細胞中這一磷酸化水平在各濃度下均顯著高于野生型,尤其在200 nM魚藤酮處理下差異zui為明顯。值得注意的是,總Drp1蛋白水平在兩種基因型和不同魚藤酮濃度下均無顯著變化,表明突變和毒素主要影響Drp1的磷酸化狀態而非表達量。這些結果共同揭示:LRRK2 G2019S突變增強Drp1 Ser616磷酸化,這種過度磷酸化可能干擾Drp1與STX17的正常相互作用,進而導致STX17從線粒體脫落,最終損害線粒體轉移功能。該發現闡明了從遺傳突變到分子事件再到功能損傷的關鍵通路,為后續藥物干預提供了明確靶點。
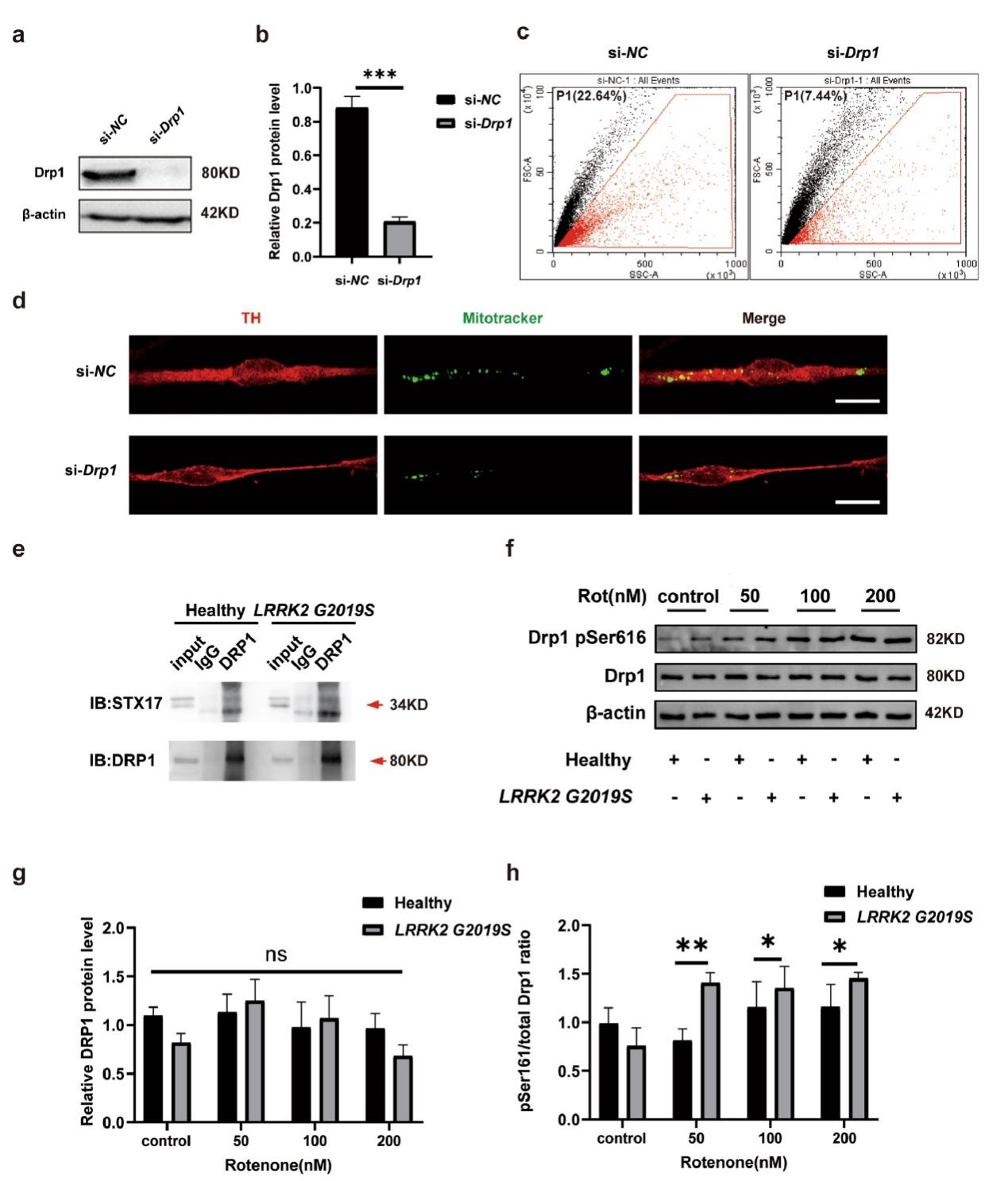
圖4,Drp1參與STX17依賴的線粒體轉移
5. LRRK2 G2019S突變與魚藤酮處理聯合影響Drp1 Ser616位點的磷酸化
在200 nM魚藤酮處理的野生型和突變型星形膠質細胞中,LRRK2抑制劑PF-06447475僅顯著降低突變型細胞的Drp1 pSer616水平,對野生型無顯著影響。相比之下,Drp1磷酸化抑制劑DUSP6在兩種基因型細胞中均能顯著降低Drp1 pSer616水平,且呈劑量依賴性。總Drp1蛋白水平在各處理組間無顯著變化。結果表明,DUSP6能有效抑制由LRRK2 G2019S突變和魚藤酮共同誘導的Drp1過度磷酸化,適合用于后續的功能挽救實驗。并比較了兩種抑制劑對Drp1 Ser616磷酸化的影響,為后續挽救實驗篩選出DUSP6。在200 nM魚藤酮處理的星形膠質細胞中,LRRK2抑制劑PF-06447475僅顯著降低突變型細胞的Drp1 pSer616水平,對野生型無顯著影響。而Drp1磷酸化抑制劑DUSP6在兩種基因型細胞中均能劑量依賴性地顯著降低Drp1 pSer616水平,總Drp1蛋白水平無變化。結果表明,DUSP6能有效抑制由LRRK2 G2019S突變和魚藤酮共同誘導的Drp1過度磷酸化,適合用于后續的功能挽救實驗。
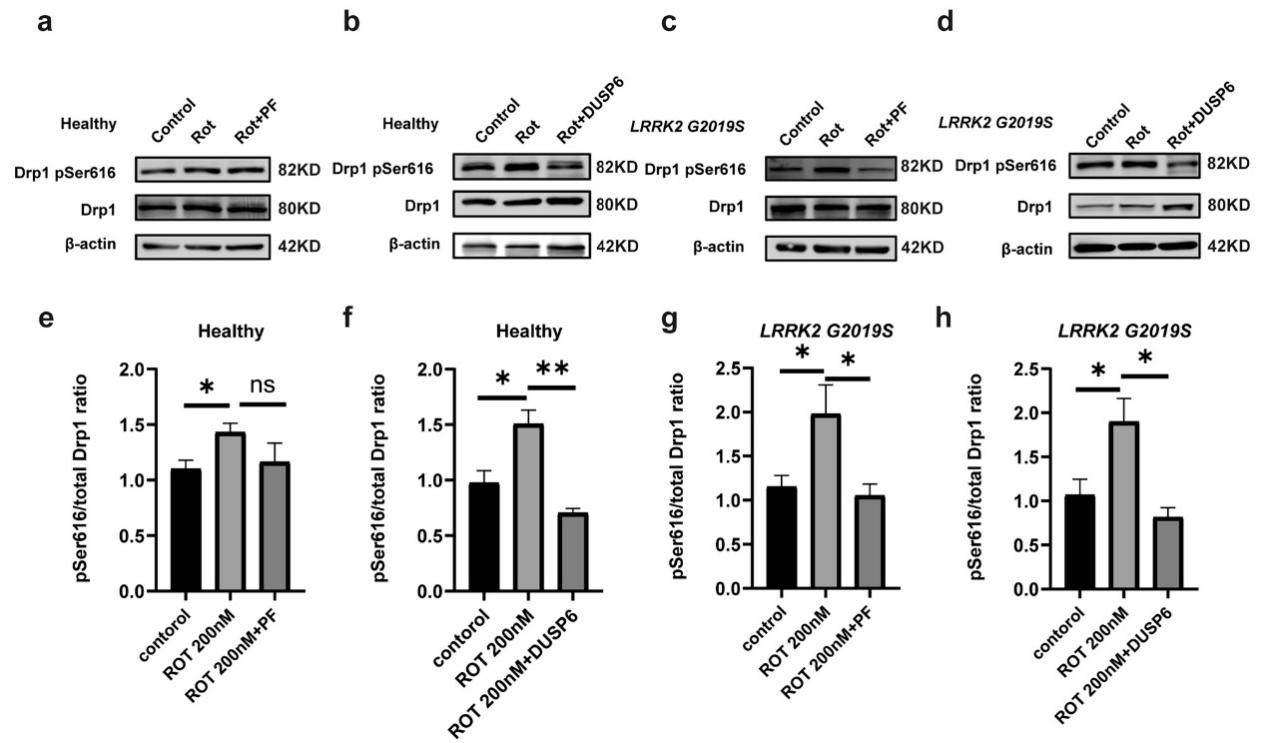
圖5,Drp1抑制劑DUSP6可降低野生型和LRRK2 G2019S突變型星形膠質細胞中Drp1 pSer616的水平
6. DUSP6挽救受損的線粒體轉移和多巴胺能神經元損傷
在200 nM魚藤酮處理下,STX17與TOM20共定位減少(野生型降37.7%,突變型降46%),而DUSP6處理后共定位顯著增加(野生型增37%,突變型增50%)。Western blot驗證STX17蛋白水平變化,但明確其線粒體靶向能力受Drp1磷酸化調控。功能上,DUSP6使線粒體轉移效率提高(野生型增27%,突變型增53%),神經元樹突長度改善(野生型增12%,突變型增111%)。結果表明,DUSP6通過抑制Drp1 Ser616磷酸化,阻止STX17從線粒體脫落,恢復線粒體轉移功能,對突變型神經元具有顯著保護作用。
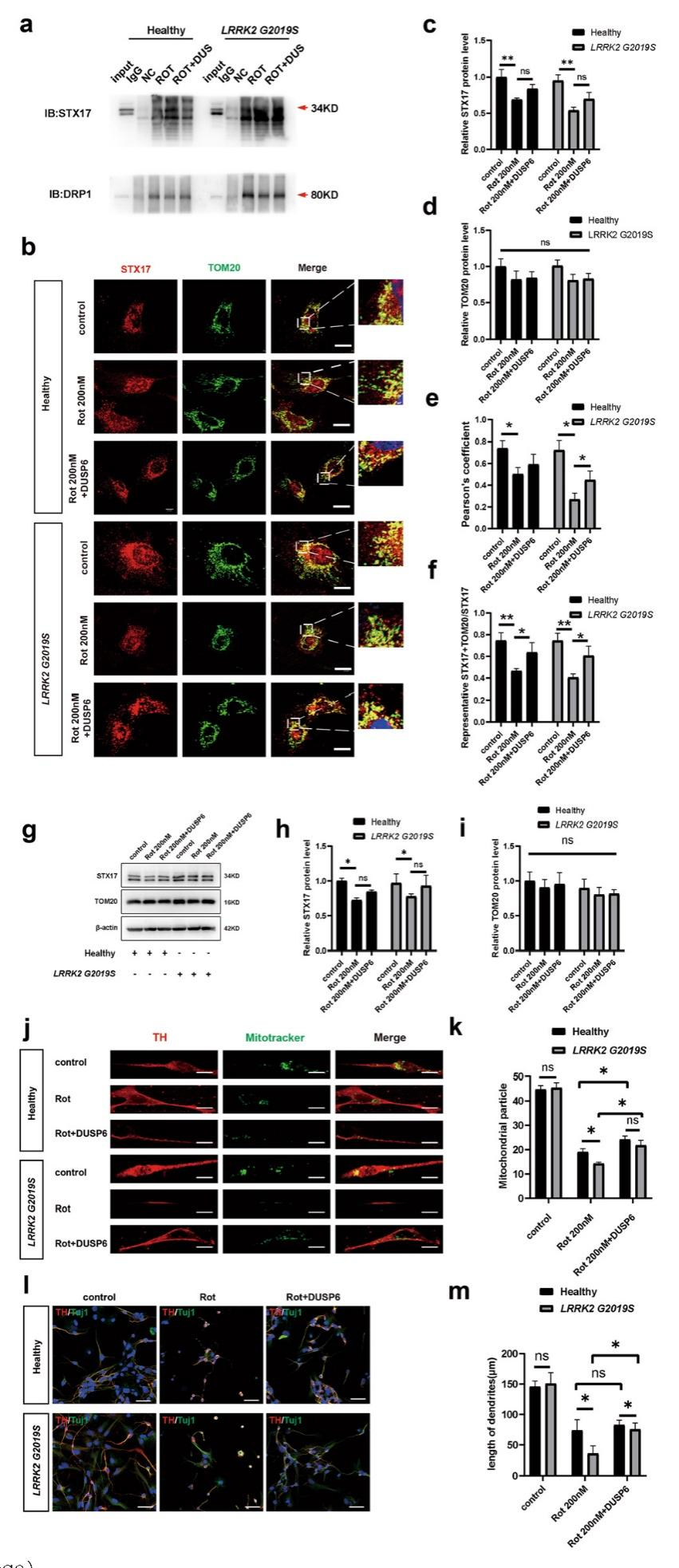
圖6,DUSP6可挽救野生型和表達LRRK2 G2019S的星形膠質細胞中受損的線粒體轉移和多巴胺能神經元損傷
7. 全文總結
研究shou次揭示LRRK2 G2019S突變通過Drp1-STX17通路損害星形膠質細胞向多巴胺能神經元的線粒體轉移功能,從而加劇環境毒素誘導的神經元損傷。研究發現,突變與魚藤酮協同增強Drp1 Ser616磷酸化,導致STX17從線粒體外膜脫落,線粒體釋放減少;而Drp1磷酸化抑制劑DUSP6可恢復STX17線粒體定位、提高線粒體轉移效率并保護神經元。并shou次將線粒體轉移功能障礙確立為LRRK2突變致帕金森病的新機制,提出Drp1-STX17軸為治療靶點,并證實DUSP6的神經保護作用。然而研究基于單例中國患者來源的iPSC,樣本量有限,且LRRK2 G2019S在中國人群罕見,研究結果向其他人群或其他LRRK2突變類型的普適性有待驗證。



